This article describes a technology to achieve broad and deep coverage of multiple molecular classes simultaneously through Multi-omics (proteome, lipidome, and metabolome) single-shot technology (MOST), requiring only one column, one LC-MS instrument, and a simplified workflow.
Mass spectrometry is the premier tool for identifying and quantifying protein phosphorylation on a global scale. Analysis of phosphopeptides requires enrichment, and even after the samples remain highly complex and exhibit a broad dynamic range of abundance. A recent publication by Muehlbauer et. al. found that incorporating a commercialized aerodynamic high-field asymmetric waveform ion mobility spectrometry (FAIMS) device into the phosphoproteomic workflow was a valuable addition with greater benefits emerging from longer analyses and higher amounts of material.
Gut microbiota can regulate host physiological and pathological status through gut–brain communications or pathways. However, the impact of the gut microbiome on the proteins involved in regulating brain functions and behaviors is still not clearly understood. In a recent publication by Liu et al., the author describes a combined label-free and 10-plex DiLeu-based quantitative method that enabled a comprehensive profiling of gut microbiome that induced dynamic changes, suggesting that the gut microbiome might mediate a range of behavioral changes, brain development, and learning and memory through these neuropeptides and proteins.
Photoactivation and photodissociation have long proven to be useful tools in tandem mass spectrometry, but implementation often involves cumbersome and potentially dangerous configurations. To remedy this problem, a fiber-optic cable was coupled to an infrared (IR) laser on a mass spectrometer. These advances allow for a more robust, straightforward, and safe instrumentation platform, permitting implementation of AI-ETD and IRMPD on commercial mass spectrometers and broadening the accessibility of these techniques.
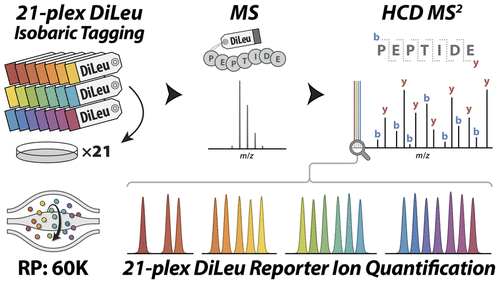
Isobaric tags enable multiplexed quantitative analysis of many biological samples in a single LC-MS/MS experiment. As a cost-effective alternative to expensive commercial isobaric tagging reagents, the lab of Lingjun Li has developed their own custom “DiLeu” isobaric tags for quantitative proteomics. In this paper, Dustin Frost showcases a new generation of DiLeu tags that achieves 21-plex quantification in high-resolution HCD MS/MS spectra.
A recent publication in Analytical Chemistry by Trenton Peters-Clarke et.al explores the promise of modified oligonucleotides for drug development, with small interfering RNAs (siRNA) and microRNAs gaining traction in the therapeutic market. Mass spectrometry (MS)-based analysis offers many benefits for characterizing modified nucleic acids. Negative electron transfer dissociation (NETD) has proven valuable in sequencing oligonucleotide anions, particularly because it can retain modifications while generating sequence-informative fragments.
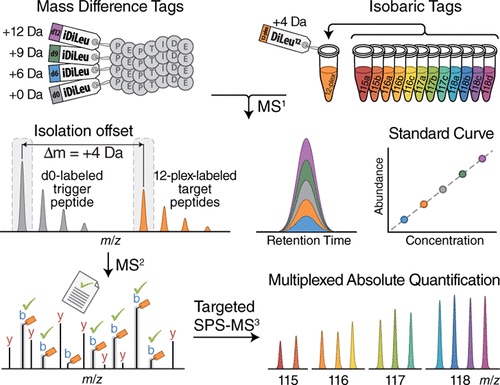
Absolute quantification is both an effective technique– which allows for robust results in proteomics research– and a challenging one. Problems that absolute quantification presents include low specificity in complex backgrounds, limited analytical throughput and wide dynamic range.
To solve these issues, Zhong et al (2019) developed hybrid offset-triggered multiplex absolute quantification (HOTMAQ), a strategy which increases the analytical throughput (the increase in analysis production rate) of targeted quantitative proteomics by up to 12 times. This technique accomplishes this by using mass-difference and isobaric tags to create an internal standard curve in the MS1 precursor scan, identify peptides at the MS2 level, and mass offset-trigger the quantification of target proteins in synchronous precursor selection at the MS3 level. All of this is accomplished at the same time.
Because HOTMAQ results in greater quantitative performance, higher flexibility and quicker analysis rate, HOTMAQ is a strategy that can easily be applied to target peptidomics, proteomics, and phosphoproteomics.
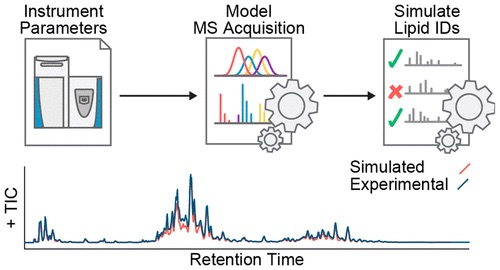
The issue of effectively profiling the diversity and range of biomolecules is an important one to consider in Mass Spectrometry, and relies on well-sought out selection of acquisition parameters. However, acquisition parameters are generally selected in a way that is time-consuming and tends to produce lacking results.
By creating an algorithm which simulates LC-MS/MS lipidomic data acquisition performance in a benchtop quadrupole-Orbitrap Mass Spectrometer system and pairing it with an algorithm that defines constrained parameter optimization, researchers were able to efficiently identify LC-MS/MS method parameter sets for specific sample matrices. Additionally, researchers used a simulation called in silico to demonstrate how developments in mass spectrometer speed and sensitivity will result in even more effective biomolecule identification.
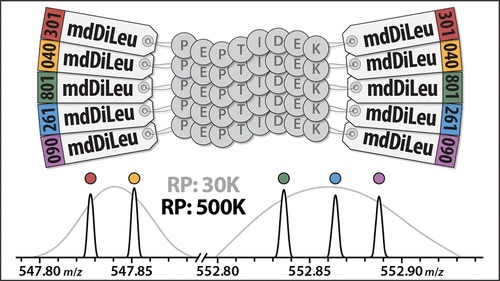
Specifically, researchers developed 5-plex mass defect N, N-dimethyl leucine (mdDiLeu) tags. These tags have multiple benefits; they can aid in the quantification of biological samples and have increased multiplexing due to the addition of mass difference isotopologues. Additionally, the synthesis of these cost effective tags is straightforward and only requires one reaction step, which can be done in any lab. Also, this mass defect-based labelling strategy is more accurate than isobaric label-based reporter ion quantification, as the latter is impacted by ratio compression.
In this paper, Zhong et al (2019) demonstrate the efficacy of 5-plex mdDiLeu tags for quantitative proteomics by conducting mass spectrometry experiments with these tags on labelled Saccharomyces cerevisiae lysate digest.
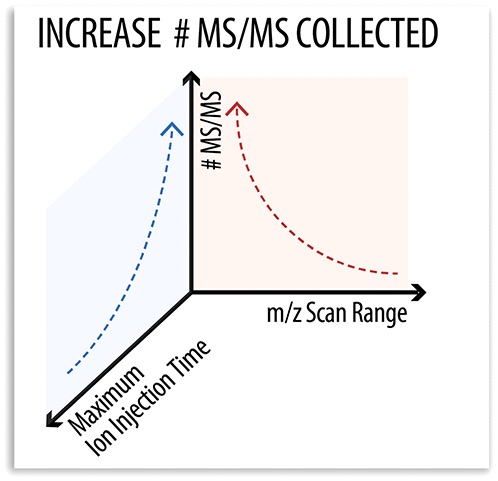
While advances in mass spectrometry (MS/MS) have lead to increased performance in shotgun proteomics experiments, ion trap scan duration is highly variable and often depends on the mass of the precursor.
Looking into this variability, the authors compared the performance of various static mass-to-charge ratio scan ranges for ion trap MS/MS acquisition to conventional dynamic mass-to-charge ratio scan ranges. Compared to the standard dynamic approach, the fixed mass-to-charge ratio scan range generated 12% more MS/MS scans and identified more unique peptides.