Olga Vitek’s tutorial lecture on Data Analysis at the first annual NAMSS
Olga Vitek from Northeastern University delivered a tutorial lecture on Data Analysis at the NCQBCS first annual North American Mass Spectrometry Summer School in 2018.
Olga Vitek from Northeastern University delivered a tutorial lecture on Data Analysis at the NCQBCS first annual North American Mass Spectrometry Summer School in 2018.
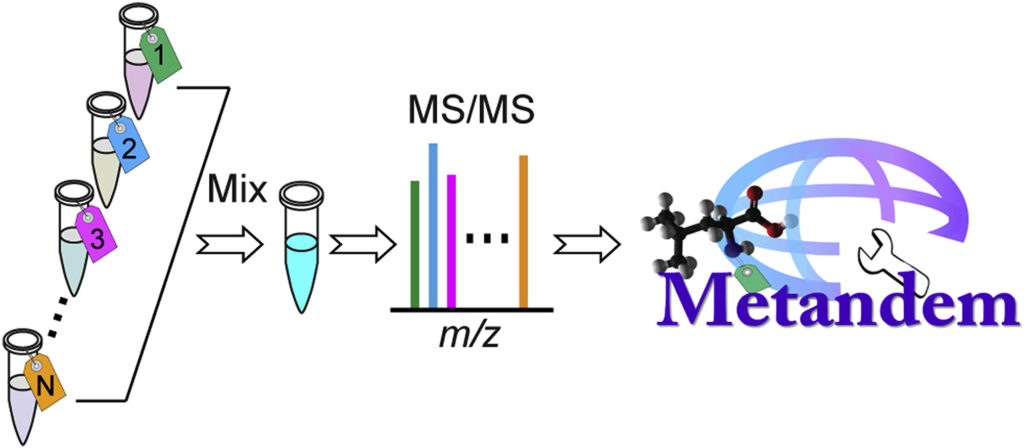
Hao et al. (2019) recently published a paper in Analytica Chimica Acta detailing the utility of Metandem, a data analysis software which is aids in isobaric labeling-based metabolomics.
While mass spectrometry-based stable isotope labeling is advantageous compared to other methods of isotope labeling due to its multiplexing and accurate quantification capabilities, its data analysis requires specifically customized bioinformatic tools. However, Metandem, a free, unique and online software, can aid in the analysis of stable isotope labeling-based metabolomics data.
Metandem has a number of different features that assist in MS-based isobaric labeling, such as integrating feature extraction, metabolite quantification and identification, batch processing of multiple data files, online parameter optimization for custom datasets, data normalization and statistical analysis.
Metatandem is available free and online at http://metandem.com/web/.
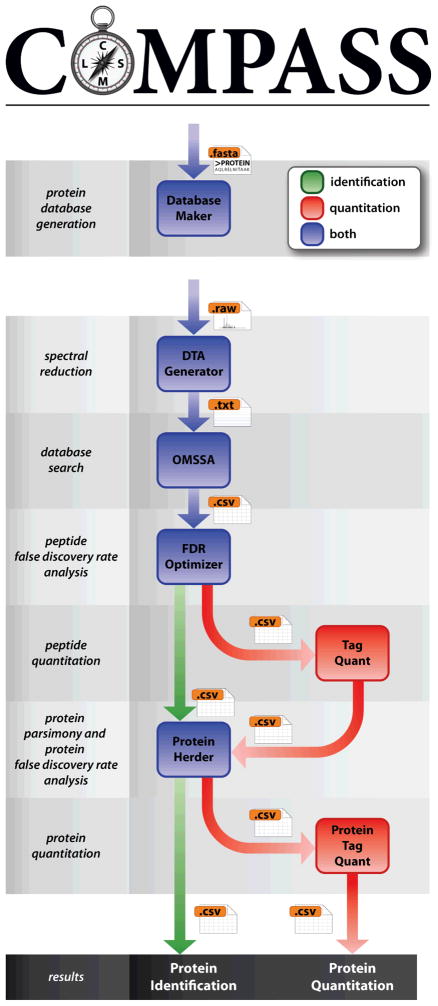
Compass is a free and open-source software pipeline designed around the Open Mass Spectrometry Search Algorithm. Compass aids in high-throughput analysis of proteomics data such as FASTA database creation, peptide-spectral matching, calculation of false discovery rates, and protein grouping, as well as spectral reduction, peptide quantitation via isobaric labeling (or without), and protein parsimony.
Furthermore Compass utilizes graphical user interfaces which work well with data files in original instrument vendor format, making it easy to use.
The manuscript for Compass is available here, and the software can be downloaded here. Additionally, information on other software that the National Center for Quantitative Biology of Complex Systems offers can be found here.
Buchberger AR et al (2019) recently published a chapter reviewing various labelling strategies for quantitative proteomic analysis in Mass Spectrometry-Based Chemical Proteomics.
Specifically, the chapter reviews strategies such as label-free quantitation, metabolic labeling, and chemical stable isotope labeling, and also discusses which labeling approach is best for various types of proteomic analyses. The chapter also provides an explanation on how to use N,N‐dimethyl alanine (DiAla) and N,N‐ dimethyl valine (DiVal) isobaric labeling strategies for quantitative analyses in ways which are economic and effective.
The chapter states that quantitative proteomics is crucial for biomarker discovery in studying and understanding various diseases and biological research, as proteins are crucial in all biological processes. Because biomarker studies can be time-consuming, heavily reliant on instruments and vary depending on the strategy used, selecting the appropriate labeling strategy is important in quantitative analysis.
NCQBCS, which works to develop next-generation protein measurement technologies for biomedical application, has programs available for a wide range of students. This means that there are introductory training programs available for those interested in learning the basics of mass spectrometry, as well as programs geared for experts on specific technologies.
NCQBCS divides its training topics into four broad categories: Sample Preparation, Instrumentation, Data Analysis, and Protein Quantification. Trainees can build their own syllabus of workshops from a variety of categories and experience levels.
Comprehensively, we offer programs in:
Sample Preparation: Peptide Fractionation, Protein Digestion, Protein extraction.
Mass Spectrometry: MS Methods, Instrument Troubleshooting, Nano-chromatography.
Data Analysis: Data Visualization, Data Interpretation, Data Searching.
Protein Quantification: Label-free, Metabolic labeling, Isobaric chemical labeling.
More information on our training programs are located here, and one can sign up for training here.
Additionally, one can also receive coaching at the 3rd Annual North American Mass Spectrometry Summer School, which will take place June 15-18, 2020. This event, which will be hosted by international experts on Mass Spectrometry, will feature workshops, lectures and networking, among other activities.
One may find more information, as well as sign up for summer school, here.
Andy Tao from Purdue University delivered a tutorial lecture on Protein PTMs at the NCQBCS first annual North American Mass Spectrometry Summer School in 2018.
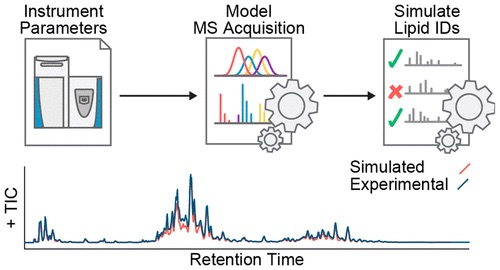
The issue of effectively profiling the diversity and range of biomolecules is an important one to consider in Mass Spectrometry, and relies on well-sought out selection of acquisition parameters. However, acquisition parameters are generally selected in a way that is time-consuming and tends to produce lacking results.
By creating an algorithm which simulates LC-MS/MS lipidomic data acquisition performance in a benchtop quadrupole-Orbitrap Mass Spectrometer system and pairing it with an algorithm that defines constrained parameter optimization, researchers were able to efficiently identify LC-MS/MS method parameter sets for specific sample matrices. Additionally, researchers used a simulation called in silico to demonstrate how developments in mass spectrometer speed and sensitivity will result in even more effective biomolecule identification.
Alzheimer’s disease begins with a long, hard-to-discern and symptom-free phase which may be a key opportunity for early diagnosis and therapeutic intervention. Zhong et al (2019) defined reliable and valid biomarkers that could identify the disease during this period, as published in a recent article in Frontiers in Molecular Neuroscience.
Alzhiemer’s disease is a progressive neurodegenerative disease which is characterized by the progressive buildup of senile plaques, neurofibrillary tangles, and loss of synapses and neurons in the brain. Behaviorally, this is presented as a progressive degeneration of overall function, such as difficulty with memory, mood instability and loss of motor function. Currently, there is no cure.
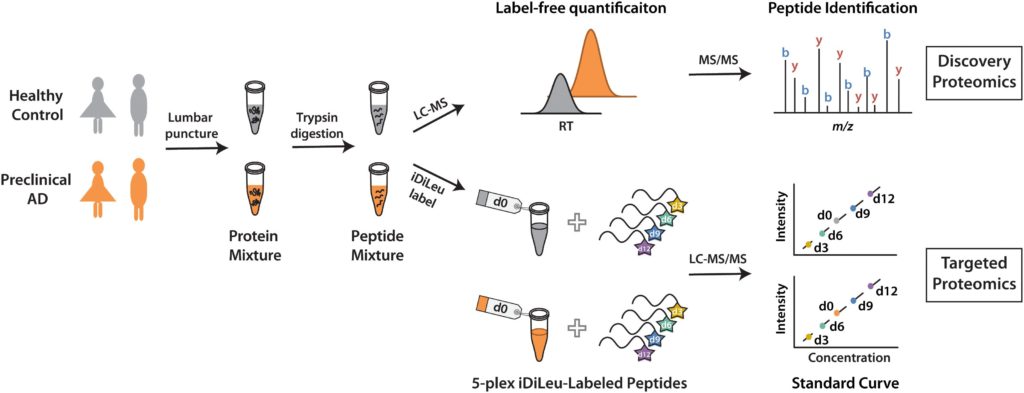
Using discover proteomics analysis of cerebrospinal fluid (CSF), Zhong et al found that in both healthy controls and in preclinical Alzheimer’s Disease patients, 732 proteins in women and 704 men proteins in men had more than one unique peptide. Then, Zhong et al found that 79 (women) and 98 (men) proteins were significantly altered in preclinical alzheimer’s patients who have already demonstrated some symptoms of mild cognitive impairment or dementia.
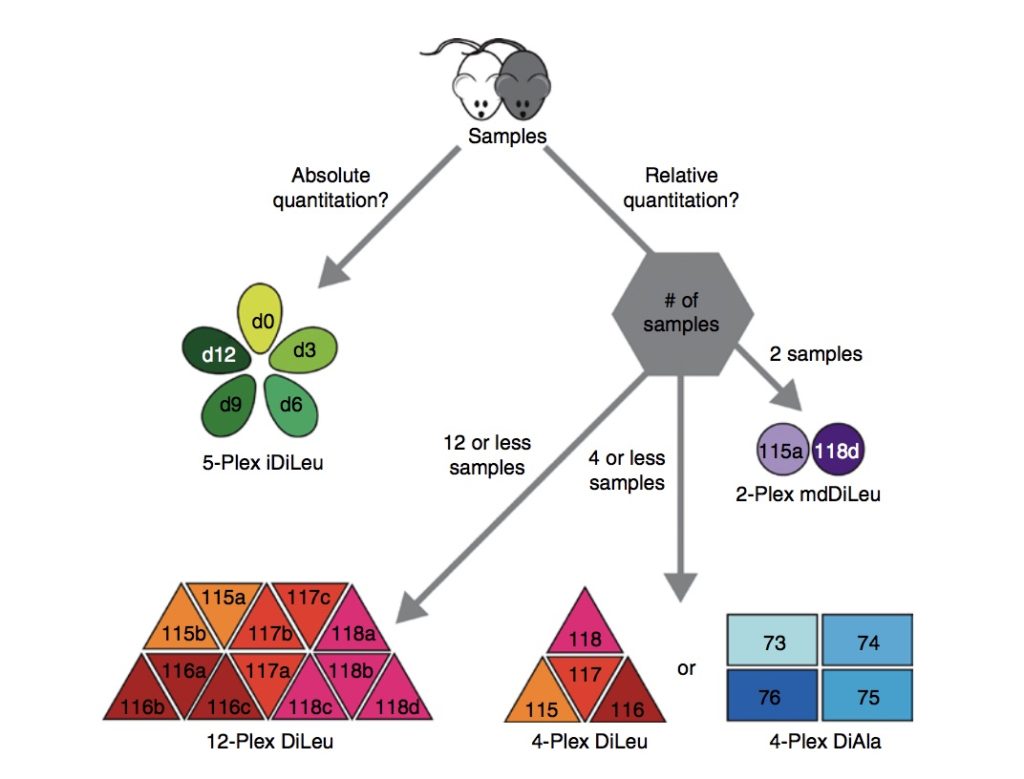
Using N,N-dimethyl leucine (iDiLeu) tags, researchers verified the Alzheimer’s disease biomarkers called neurosecretory protein VGF and apolipoprotein E. Then, researchers used a four-point internal calibration curve to determine the “absolute amount” of target analytes in cerebrospinal fluid through a single liquid chromatography-mass spectrometry run.
Ma et al (2019) recently published a paper on the quantification of human pancreatic extracellular matrix proteins in the Journal of Proteome Research.
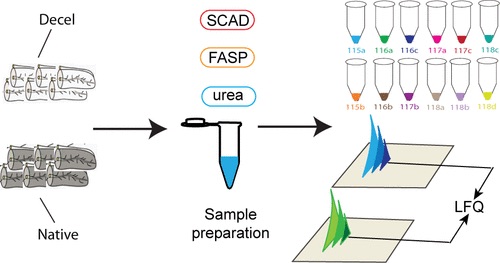
In this study, researchers characterized the composition of the human pancreatic extracellular matrix (ECM) before and after decellularization. To find the relative quantification of ECM proteins, they used isobaric dimethylated leucine (DiLeu) labeling.
It was important for researchers to look at the ECM of the pancreatic microenvironment as it is essential to pancreatic function– it regulates β cell proliferation, differentiation, and insulin secretion.
As a result of decellularization, and through quantitative proteomic analysis, most cellular proteins were removed while matrisome proteins remained. This process generated a large data set of matrisome proteins from a single tissue type.
Researchers then quantified the distinct expression of ECM proteins, comparing adult and fetal pancreas ECM. This revealed a correlation between matrix composition and postnatal β cell maturation.
Overall, the results of this study sheds light on the prospect of bioengineering a pancreas. Additionally, the study demonstrates the roles that matrisome proteins have in postnatal β cell maturation.
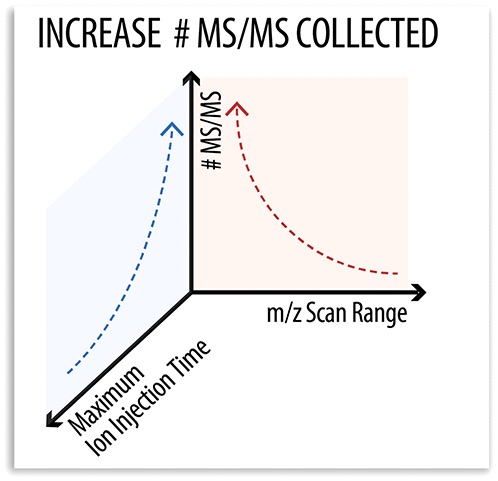
Trujillo et al (2019) published an article on maximizing tandem mass spectrometry acquisition rates for shotgun proteomics in a recent issue of Analytical Chemistry.
While advances in mass spectrometry (MS/MS) have lead to increased performance in shotgun proteomics experiments, ion trap scan duration is highly variable and often depends on the mass of the precursor.
Looking into this variability, the authors compared the performance of various static mass-to-charge ratio scan ranges for ion trap MS/MS acquisition to conventional dynamic mass-to-charge ratio scan ranges. Compared to the standard dynamic approach, the fixed mass-to-charge ratio scan range generated 12% more MS/MS scans and identified more unique peptides.