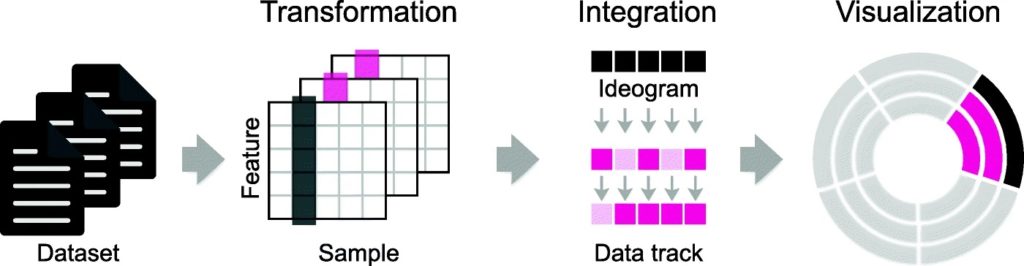
MS-Helios is an easy-to-use command line tool which works to solve the challenge of data analysis and visualization in the face of high-resolution mass spectrometery.
Though high-resolution mass spectrometry can identify hundreds of metabolites and thousands of proteins, this can make data analysis and visualization hard to do.
MS-Helios is a solution, allowing for compact data representation and reduced dimensionality. This tool also allows non-experts and experts alike to generate data and configuration files and publish high-quality, circular plots with Circos.
This software is available for download here. The manuscript for MS-Helios can be viewed here.
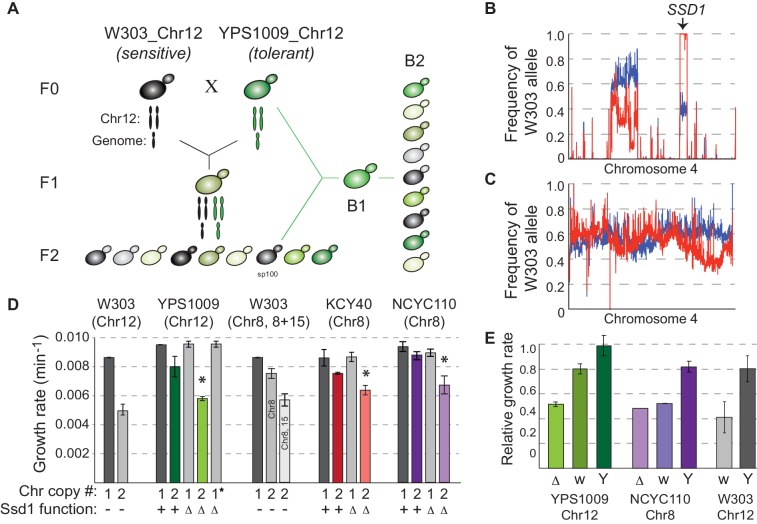
Hose et al (2020) researched the genetic basis of aneuploidy tolerance in wild yeast in a recent edition of Elife.
Aneuploidy, or the abnormal chromosome numbers in a cell, is a harmful condition in developmental stages of wild yeast, yet is also common in plant cancers and pathogenic fungi. It is interesting to note that aneuploidy tolerance varies; for instance, researchers found that some wild isolates of baker’s yeast can tolerate chromosome amplification, while laboratory strains cannot.
To study the genetic basis of how well wild yeast can tolerate aneuploidy, researchers mapped the genetic basis to Ssd1, an RNA-binding translational regulator that functions in wild strains but is defective in a laboratory strain “W303.”
Researchers found that aneuploidy tolerance is enabled via a role for Ssd1 in mitochondrial physiology, such as binding and regulating nuclear-encoded mitochondrial mRNAs.
Patients with chronic obstructive pulmonary disease (COPD), a disease that is characterized by obstructed airflow to the lungs and is associated with long-term exposure to cigarette smoke, often also develop skeletal muscle dysfunction. Overall, the comorbidity of COPD and skeletal muscle dysfunction is associated with outcomes such as poor health and mortality.
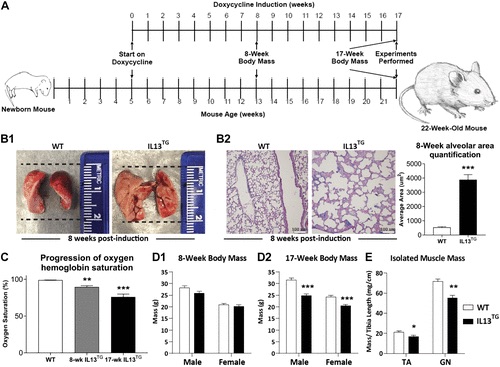
While some research has suggested that skeletal muscle dysfunction may be the result of COPD-related conditions, such as protein degradation and metabolic disruption, there is still poor understanding on what mechanisms would regulate these processes, as there is little to no research on a validated animal model of pulmonary emphysema. Therefore, Balnis et al (2020) sought to use such a model based on inducible UL-13-driven pulmonary emphysema (IL-13TG) to study the mechanisms of skeletal muscle dysfunction.
Using a transgenic mouse model, researchers found that the skeletal muscles of emphysematous mice are similar to those developed by human patients with COPD. For instance, both groups develop muscular atrophy and have decreased oxygen consumption. Within skeletal muscles, both groups also had downregulated ATP binding and bioenergetics.
Researchers concluded that transgenic animal models of COPD are useful to understand skeletal-muscle dysfunction in humans.
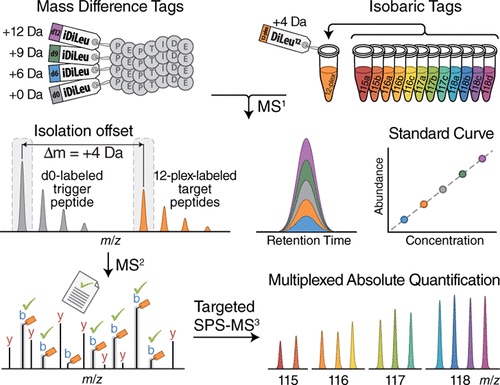
Absolute quantification is both an effective technique– which allows for robust results in proteomics research– and a challenging one. Problems that absolute quantification presents include low specificity in complex backgrounds, limited analytical throughput and wide dynamic range.
To solve these issues, Zhong et al (2019) developed hybrid offset-triggered multiplex absolute quantification (HOTMAQ), a strategy which increases the analytical throughput (the increase in analysis production rate) of targeted quantitative proteomics by up to 12 times. This technique accomplishes this by using mass-difference and isobaric tags to create an internal standard curve in the MS1 precursor scan, identify peptides at the MS2 level, and mass offset-trigger the quantification of target proteins in synchronous precursor selection at the MS3 level. All of this is accomplished at the same time.
Because HOTMAQ results in greater quantitative performance, higher flexibility and quicker analysis rate, HOTMAQ is a strategy that can easily be applied to target peptidomics, proteomics, and phosphoproteomics.
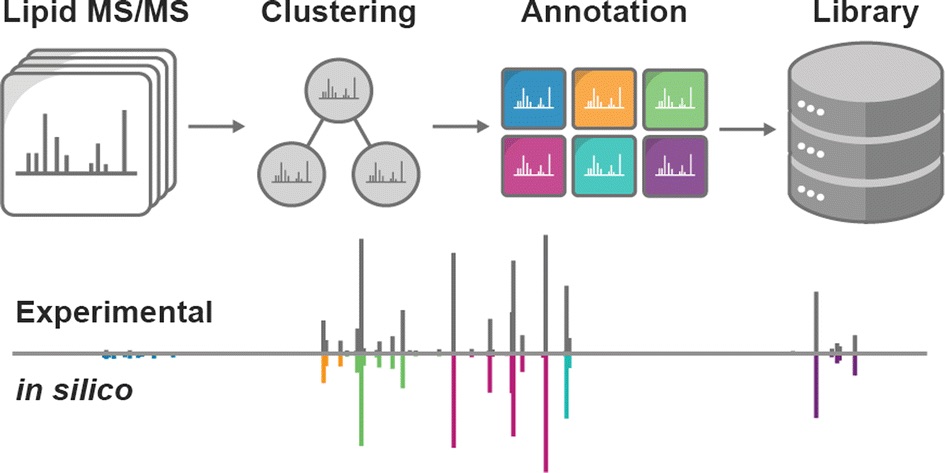
LipiDex is a free and open-source software package offered by NCQBCS. This software package unifies all stages of the LC-MS/MS lipid identification process, and also utilizes intelligent data filtering to reduce manual result curation while increasing identification confidence.
One can use LipiDex to accomplish a variety of functions. For instance, one can create and manage custom in-silico lipid spectral libraries; model complex lipid MS/MS fragmentation using intuitive fragmentation templates; generate high-confidence MS/MS lipid identifications; annotate chromatographic peak tables with lipid identifications; and automatically filter peak tables for adduct peaks, in-source fragments and dimers.
Information on both LipiDex and Library Forge can be found here, and the software download is located here. Additionally, information on other software that the National Center for Quantitative Biology of Complex Systems offers can be found here.
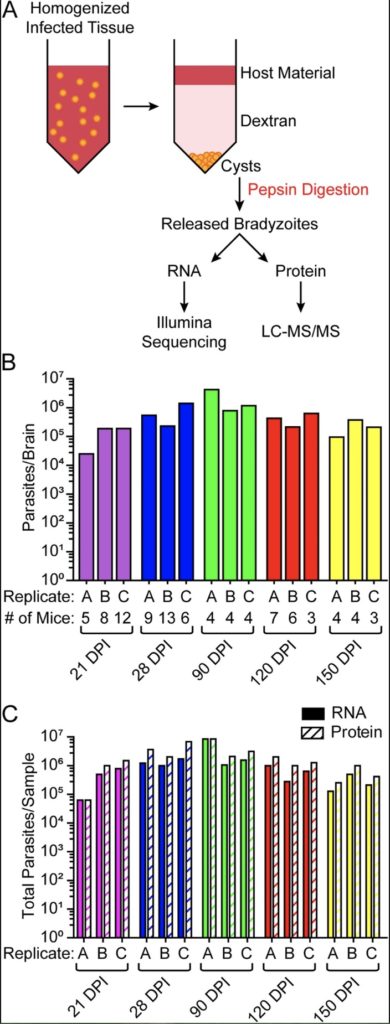
Toxoplasma gondii is a protozoan pathogen responsible for the infectious disease “toxoplasmosi,” and this pathogen is of researcher utility as it is capable of infecting a host’s brain, transitioning from fast-growing to latent morphology morphology life stages (from “tachysoite” to “bradyzoite”), and eventually creating neuronal cysts which are largely invisible to the host, as well as resilient against the host’s immune response and modern therapeutics.
Garfoot et al analyzed results from transcriptional and proteomic analyses of fast-growing (bradyzoite) fractions of the infection from mouse brains over a period of 21-150 days and, through deep sequencing of expressed transcripts found that one third of the transcripts were more enriched compared to the slow-growing tachysoites. Furthermore, researchers found that the transcript which grew the most over the course of the infection was the sporoAMA1 transcript.
As a result of this work, researchers have expanded the transcriptional profile of in vivo toxoplasmosis bradyzoites.
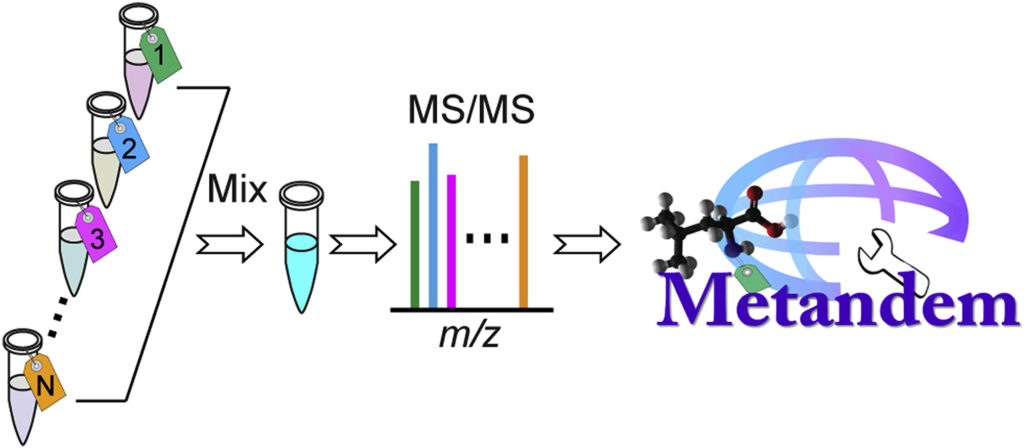
While mass spectrometry-based stable isotope labeling is advantageous compared to other methods of isotope labeling due to its multiplexing and accurate quantification capabilities, its data analysis requires specifically customized bioinformatic tools. However, Metandem, a free, unique and online software, can aid in the analysis of stable isotope labeling-based metabolomics data.
Metandem has a number of different features that assist in MS-based isobaric labeling, such as integrating feature extraction, metabolite quantification and identification, batch processing of multiple data files, online parameter optimization for custom datasets, data normalization and statistical analysis.
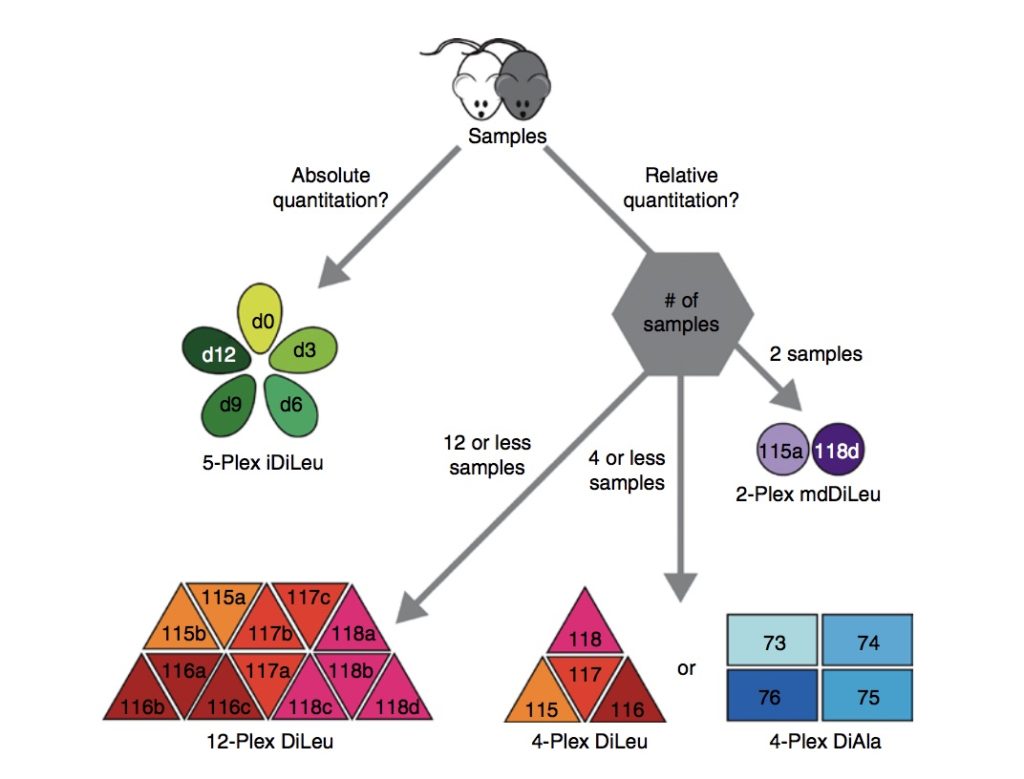
Specifically, the chapter reviews strategies such as label-free quantitation, metabolic labeling, and chemical stable isotope labeling, and also discusses which labeling approach is best for various types of proteomic analyses. The chapter also provides an explanation on how to use N,N‐dimethyl alanine (DiAla) and N,N‐ dimethyl valine (DiVal) isobaric labeling strategies for quantitative analyses in ways which are economic and effective.
The chapter states that quantitative proteomics is crucial for biomarker discovery in studying and understanding various diseases and biological research, as proteins are crucial in all biological processes. Because biomarker studies can be time-consuming, heavily reliant on instruments and vary depending on the strategy used, selecting the appropriate labeling strategy is important in quantitative analysis.
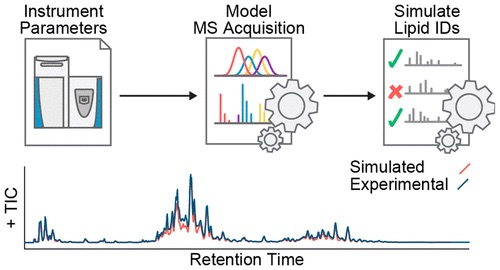
The issue of effectively profiling the diversity and range of biomolecules is an important one to consider in Mass Spectrometry, and relies on well-sought out selection of acquisition parameters. However, acquisition parameters are generally selected in a way that is time-consuming and tends to produce lacking results.
By creating an algorithm which simulates LC-MS/MS lipidomic data acquisition performance in a benchtop quadrupole-Orbitrap Mass Spectrometer system and pairing it with an algorithm that defines constrained parameter optimization, researchers were able to efficiently identify LC-MS/MS method parameter sets for specific sample matrices. Additionally, researchers used a simulation called in silico to demonstrate how developments in mass spectrometer speed and sensitivity will result in even more effective biomolecule identification.
Alzhiemer’s disease is a progressive neurodegenerative disease which is characterized by the progressive buildup of senile plaques, neurofibrillary tangles, and loss of synapses and neurons in the brain. Behaviorally, this is presented as a progressive degeneration of overall function, such as difficulty with memory, mood instability and loss of motor function. Currently, there is no cure.
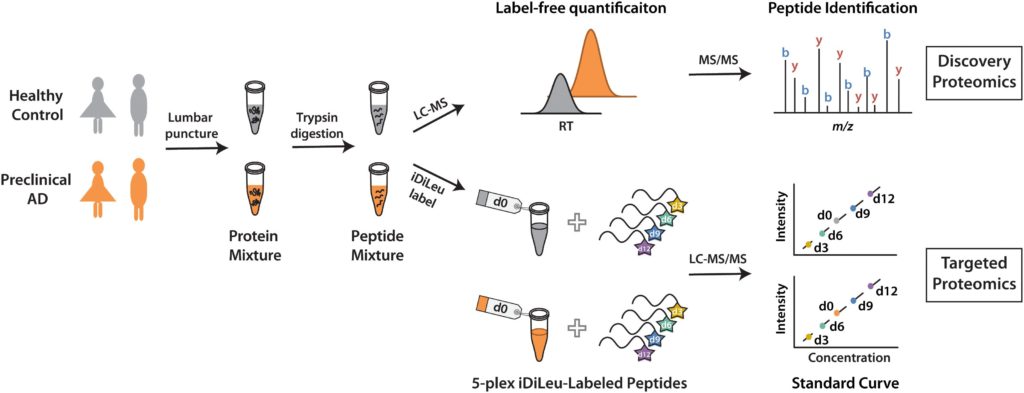
Using discover proteomics analysis of cerebrospinal fluid (CSF), Zhong et al found that in both healthy controls and in preclinical Alzheimer’s Disease patients, 732 proteins in women and 704 men proteins in men had more than one unique peptide. Then, Zhong et al found that 79 (women) and 98 (men) proteins were significantly altered in preclinical alzheimer’s patients who have already demonstrated some symptoms of mild cognitive impairment or dementia.
Using N,N-dimethyl leucine (iDiLeu) tags, researchers verified the Alzheimer’s disease biomarkers called neurosecretory protein VGF and apolipoprotein E. Then, researchers used a four-point internal calibration curve to determine the “absolute amount” of target analytes in cerebrospinal fluid through a single liquid chromatography-mass spectrometry run.