A Strategy for Increasing Analytical Throughput in Quantitative Proteomics
Zhong et al (2019) developed a novel strategy aimed towards solving challenges in absolute quantification, and detailed these efforts in a recent issue of Analytical Chemistry.
Absolute quantification is both an effective technique– which allows for robust results in proteomics research– and a challenging one. Problems that absolute quantification presents include low specificity in complex backgrounds, limited analytical throughput and wide dynamic range.
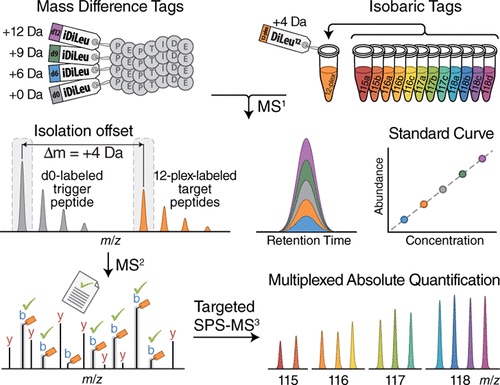
To solve these issues, Zhong et al (2019) developed hybrid offset-triggered multiplex absolute quantification (HOTMAQ), a strategy which increases the analytical throughput (the increase in analysis production rate) of targeted quantitative proteomics by up to 12 times. This technique accomplishes this by using mass-difference and isobaric tags to create an internal standard curve in the MS1 precursor scan, identify peptides at the MS2 level, and mass offset-trigger the quantification of target proteins in synchronous precursor selection at the MS3 level. All of this is accomplished at the same time.
Because HOTMAQ results in greater quantitative performance, higher flexibility and quicker analysis rate, HOTMAQ is a strategy that can easily be applied to target peptidomics, proteomics, and phosphoproteomics.