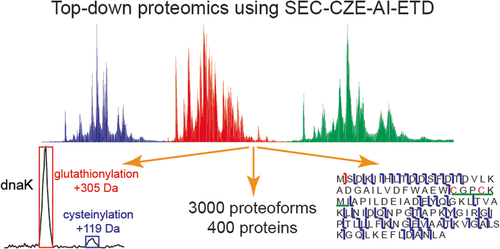
Recent research shows activated ion electron transfer dissociation has better performance for proteoform fragmentation
Elijah McCool, a graduate student in Lab of Dr. Liangliang Sun at Michigan State University, recently published on a collaboration with NCBBCS, Capillary Zone Electrophoresis-Tandem Mass Spectrometry with Activated Ion Electron Transfer Dissociation for Large-scale Top-down Proteomics. in the Journal of The American Society for Mass Spectrometry.
Capillary zone electrophoresis-tandem mass spectrometry is recognized as an efficient approach for top-down proteomics because of its high-capacity separation and highly sensitive detection of proteoforms. However, the commonly used collision-based methods often don’t provide the extensive fragmentation needed for thorough characterization of proteoforms. Activated ion electron transfer dissociation (AI-ETD), which combines infrared photoactivation with ETD, has shown better performance for proteoform fragmentation than other methods.