MS analyses profiles neuropeptides to understand their role in biology
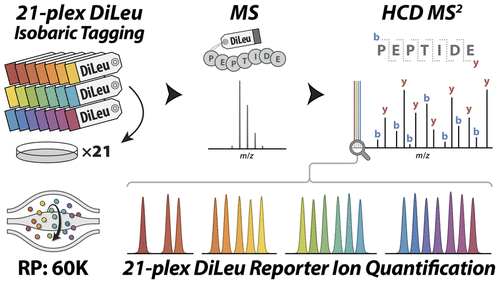
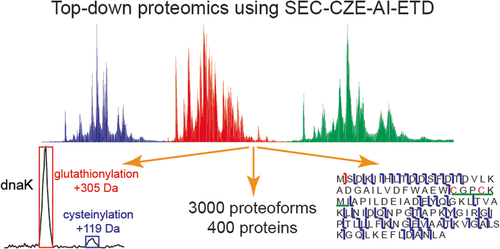
This paper by Sauer et. al. is focused on developments in mass spectrometry for labeling strategies, post-translational modification analysis, mass spectrometry imaging, and integrated multi-omic workflows, with discussion emphasizing quantitative advancements.
Read the article: Developing mass spectrometry for the quantitative analysis of neuropeptides.