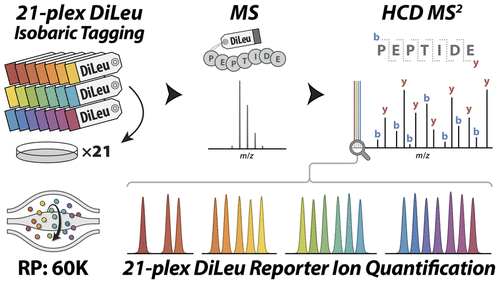
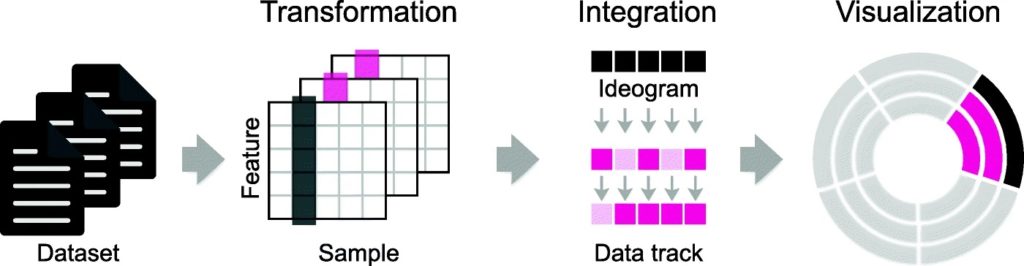
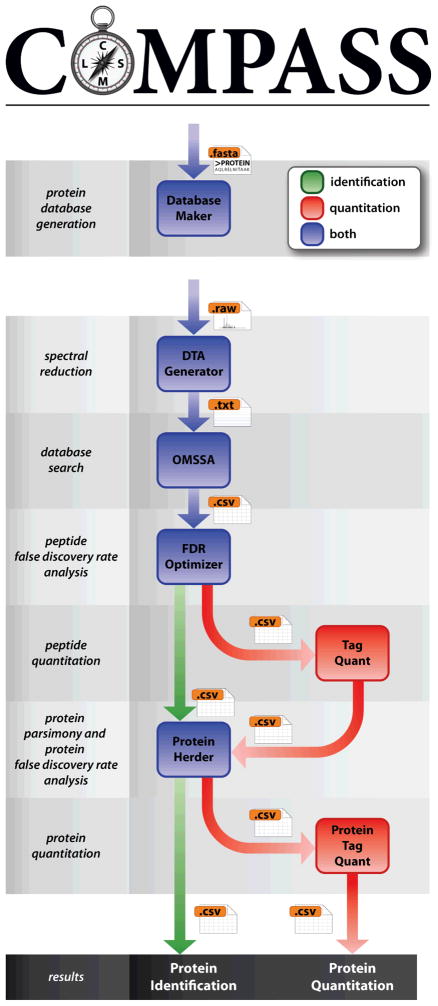
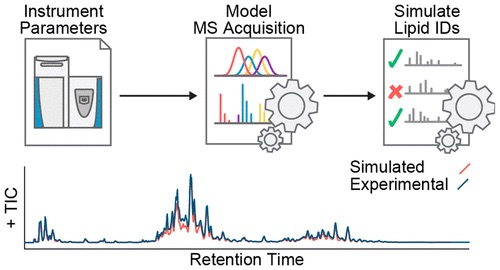
A key goal of the National Center for Quantitative Biology of Complex Systems is to extend its expertise to the broader scientific community. Therefore, NCQBCS offers hands-on-training programs ranging from basic basic proteomic methodology to advanced technological techniques.
NCQBCS, which works to develop next-generation protein measurement technologies for biomedical application, has programs available for a wide range of students. This means that there are introductory training programs available for those interested in learning the basics of mass spectrometry, as well as programs geared for experts on specific technologies.
NCQBCS divides its training topics into four broad categories: Sample Preparation, Instrumentation, Data Analysis, and Protein Quantification. Trainees can build their own syllabus of workshops from a variety of categories and experience levels.
Comprehensively, we offer programs in:
Sample Preparation: Peptide Fractionation, Protein Digestion, Protein extraction.
Mass Spectrometry: MS Methods, Instrument Troubleshooting, Nano-chromatography.
Data Analysis: Data Visualization, Data Interpretation, Data Searching.
Protein Quantification: Label-free, Metabolic labeling, Isobaric chemical labeling.
More information on our training programs are located here, and one can sign up for training here.
Additionally, one can also receive coaching at the 3rd Annual North American Mass Spectrometry Summer School, which will take place June 15-18, 2020. This event, which will be hosted by international experts on Mass Spectrometry, will feature workshops, lectures and networking, among other activities.
One may find more information, as well as sign up for summer school, here.