Selecting a Labeling Strategy for Quantitative Proteomics of Multiple Samples
Buchberger AR et al (2019) recently published a chapter reviewing various labelling strategies for quantitative proteomic analysis in Mass Spectrometry-Based Chemical Proteomics.
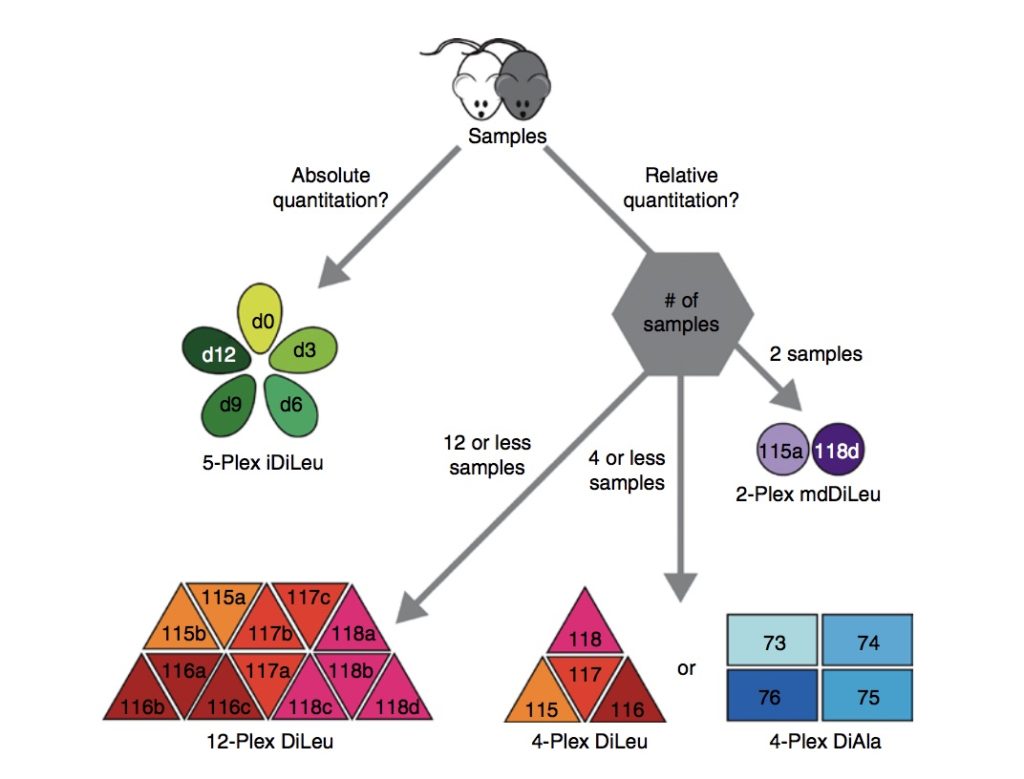
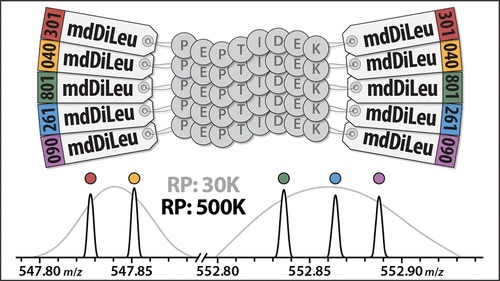
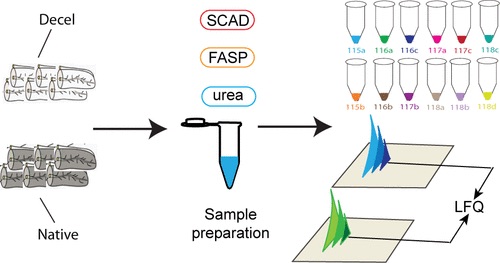
Specifically, the chapter reviews strategies such as label-free quantitation, metabolic labeling, and chemical stable isotope labeling, and also discusses which labeling approach is best for various types of proteomic analyses. The chapter also provides an explanation on how to use N,N‐dimethyl alanine (DiAla) and N,N‐ dimethyl valine (DiVal) isobaric labeling strategies for quantitative analyses in ways which are economic and effective.
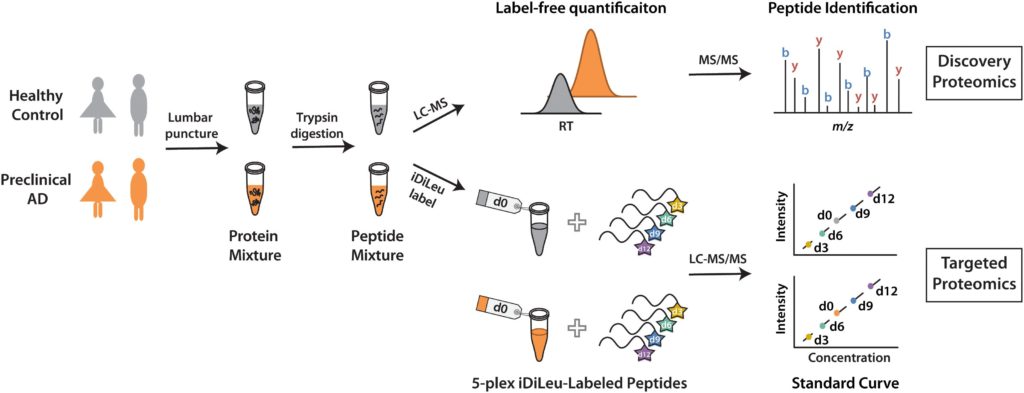
The chapter states that quantitative proteomics is crucial for biomarker discovery in studying and understanding various diseases and biological research, as proteins are crucial in all biological processes. Because biomarker studies can be time-consuming, heavily reliant on instruments and vary depending on the strategy used, selecting the appropriate labeling strategy is important in quantitative analysis.