LipidGenie, a genome-guided lipid identification tool
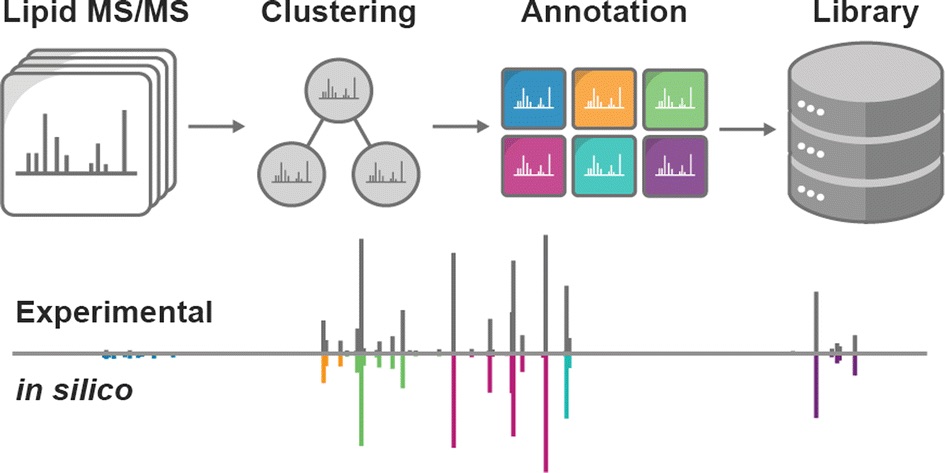
Despite the crucial roles of lipids in metabolism, we are still in the early stages of annotating lipid species and their genetic basis. To help in this work, a team of researchers led by Vanessa Linke recently developed LipidGenie, an interactive, query-able resource for lipid identification. The research team used high-resolution liquid chromatography–tandem mass spectrometry to quantify 3,283 molecular features from the liver and plasma of outbred mice. These features were then mapped to 5,622 lipid quantitative trait loci, compiled and cross-referenced to the human genome.