MS-Helios for Compact Data Visualization of Multi-omic Datasets
MS-Helios is an easy-to-use command line tool which works to solve the challenge of data analysis and visualization in the face of high-resolution mass spectrometery.
Though high-resolution mass spectrometry can identify hundreds of metabolites and thousands of proteins, this can make data analysis and visualization hard to do.
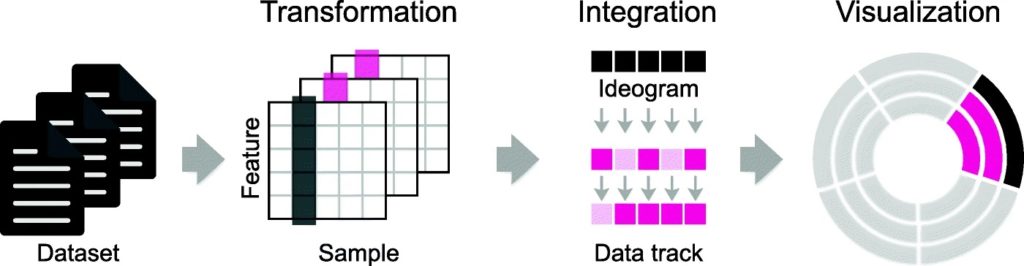
MS-Helios is a solution, allowing for compact data representation and reduced dimensionality. This tool also allows non-experts and experts alike to generate data and configuration files and publish high-quality, circular plots with Circos.
This software is available for download here.
The manuscript for MS-Helios can be viewed here.